




Термином "липиды" в химии обозначают группу различных по своим свойствам соединений, растворимых в ряде органических растворителей и нерастворимых в воде. В эту группу входят собственно жиры (глицериды) и жироподобные вещества (фосфолипиды, стерины, воски и др.). В пищевой технологии и товароведении используют термин "жир", под которым также подразумевают сумму веществ, извлекаемых органическими растворителями. При практически полном извлечении жира из пищевых продуктов термин "жир" равнозначен термину "липиды".
Дня установления количества липидов, содержащихся в пищевых продуктах, разработано несколько методов.
В целях технологического контроля за содержанием липидов в пищевых продуктах в ряде случаев применяют методы непосредственного определения содержания липидов в объектах: метод ядерного магнитного резонанса для определения содержания жира в семенах масличных культур; инфракрасную спектроскопию и турбодиметрию для определения жира в молоке и др.
Липиды пищи являются не только источниками энергии для организма, но и содержат ряд физиологически активных веществ (полиненасыщенные жирные кислоты, стерины, фосфолипиды, жирорастворимые витамины). Определения лишь общего количественного содержания липидов в продуктах питания недостаточно для полной характеристики их пищевой ценности. Таким образом, при анализе липидного состава продуктов должен быть использован комплекс методов, обеспечивающих полное извлечение липидов из продуктов, определение их количества и возможность качественной и количественной характеристики отдельных компонентов.
ИЗВЛЕЧЕНИЕ ЛИПИДОВ ИЗ ПИЩЕВЫХ ПРОДУКТОВ
Естественно, что все методы экстракции, в которых возможно изменение нативных свойств веществ, не могут быть применены. Например, использование метода Сокслета приводит к появлению значительного количества продуктов окисления, которые мешают дальнейшему проведению анализа фракционного состава и искажают его результаты.
Разнообразная природа пищевых продуктов, обусловливающая различную прочность связи липидов с другими составными частями продукта, оказывает выраженное влияние на эффективность экстракции. Ранее предложенные методы экстракции основывались главным образом на использовании неполярных растворителей (диэтиловый эфир, тетрахлорэтилен, гексан и др.). Экстракция осуществляется в специальных прибор ах-экстракторах (Сокслета, Гольдфиша, Можоннье, Фосслет, Сокстек и др.). При использовании указанных методов извлекаются главным образом свободные липиды. Прочно связанные липиды при этом не экстрагируются как из продуктов растительного, так и животного происхождения. В связи с этим, а также ввиду значительного окисления липидов в процессе выделения были предприняты поиски других, более эффективных способов экстракции. Установили, что достаточно полная экстракция липидов может бьггь осуществлена, если использовать смесь полярного растворителя и неполярного или слабополярного. Обычно используемый в качестве полярного компонента спирт ослабляет прочность комплекса липиды-белки, что обеспечивает полноту экстракции не полярным растворителем. Однако эффективность экстракции в значительной мере зависит от степени разрушения клеточной структуры исследуемых объектов. Для этого используют гидролиз, разрушение в кавитационной мельнице, измельчение продуктов, предварительно замороженных в жидком азоте.
Фольч с сотрудниками [21 ] предложил метод извлечения липидов из животных тканей смесью хлороформа и метанола в соотношении 2:1. Этим методом экстрагируются не только липиды, но и вещества нелипидной природы, растворимые в спирте. Липиды очищают от примесей с помощью фазового расслоения слабыми водными растворами сильных электролитов (например, 0,87 %-ным раствором КСI). Метод получил широкое распространение. В дальнейшем Блай и Дайер [15] модифицировали этот метод, сократив время экстракции и дав возможность получать больший выход липидов.
Кузнецов Д. И., Гришина Н. Л., Некрасова Л. В. (институт питания АМН СССР) разработали и апробировали унифицированную систему методов выделения и количественного определения липидов (УСМВОЛ) в пищевых продуктах [3]. В соответствии с этой системой в качестве экстрагирующей смеси используют хлороформ и этанол, как правило в соотношении 2:1. Экстракцию осуществляют в специальной фильтрующей делительной воронке. Определение липидов состоит из следующих методических операций: отбора и приготовления средних проб для анализа. Одну часть проб используют для определения влажности, другую – для извлечения жира (в обоих случаях берется по три повторности). Общие правила отбора и подготовки проб изложены выше в разделе "Подготовка проб к анализу". Навески продуктов животного происхождения перед экстракцией измельчают в мясорубке, растительного – в кофемолке. В результате экстракции получают сухой обезжиренный остаток и "сырой жир", т. е. сумму свободных и связанных липидов вместе с нелипидными примесями. Если необходимо выделить прочносвязанные липиды, проводят разрушение обезжиренного остатка и одновременно экстракцию в кавитационной мельнице [9]. Прочносвязанные солеобразующие липиды целесообразно выделять, проводя обычный гидролиз соляной кислотой. Из сырого жира отделяют нелипиды (эта операция обязательна при определении фракционного состава) и в результате получают сумму свободных и связанных липидов. При проведении в целях экспресс-контроля сравнительного анализа однотипных объектов с незначительными нелипидными примесями количеством нелипидов можно пренебречь и специальную очистку не проводить. Обычно одну часть экстракта используют для определения состава липидов, другую – для определения их количества. Количество липидов устанавливают взвешиванием. Для каждой группы пищевых продуктов подбирают оптимальные условия экстракции [3] .
Как отмечалось выше, для извлечения липидов из пищевых продуктов ранее широко использовались методы Сокслета [7, 10] . Эти методы в настоящее время рекомендуется применить только для продуктов, в которых преобладают триглицериды (растительные масла и подобные продукты) [10] . В остальных продуктах (зерно, мясо, и др.), где в большом количестве представлены прочносвязанные липиды, методы типа Сокслета не применимы [13] .
Для определения общего содержания жира (и только) в ряде случаев используются различные варианты щелочного или кислотного гидролиза [10]. Гидролиз осуществляют обычно путем добавления NaOH или КОН для проведения щелочного или НСI для кислотного гидролиза. Щелочные гидролизаты после окончания гидролиза подкисляют кислотой. Затем из кислогного (или подкисленного щелочного гидролизата) липиды экстрагируют гексаном или серным эфиром [17]. Более удобно пользоваться кислотным гидролизом. Следует указать, что вышеуказанными методами липиды в нативном состоянии выделить невозможно, так как в экстракт переходят в основном образовавшиеся при гидролизе жирные кислоты. Несмотря на то, что гидролитические методы во многих продуктах дают результаты, близкие к методам, основанным на использовании смеси растворителей, большинство данных по общему содержанию липидов, приведенных в настоящем справочнике, получены с использованием экстракции смесью хлороформ-метанол (по Фольчу или Блаю и Дайеру) или хлороформ-этанол (по Д. И. Кузнецову и Н. Л. Гришиной), а для масличных продуктов – методом Сокслета.
ОПРЕДЕЛЕНИЕ ФРАКЦИОННОГО СОСТАВА ЛИПИДОВ
ПИЩЕВЫХ ПРОДУКТОВ
В данной книге кроме данных об общем количестве липидов в пищевых продуктах приведены важнейшие характеристики их химического состава. Полностью отразить данные о количестве всех химических соединений, содержащихся в составе липидов, не представляется возможным. В таблицы внесены лишь те из них, которые учитываются в настоящее время при построении рационов питания.
Важнейшей особенностью, определяющей характер биологического действия пищевого жира, является состав его жирных кислот. Для удобства практического использования эти данные представлены в граммах индивидуальных жирных кислот, содержащихся в 100 г продукта. Кроме того, в таблицах приведены данные о содержании стеринов (холестерина в продуктах животного происхождения или иных стеринов в продуктах растительного происхождения), фосфолипидов. Содержание этих жироподобных веществ, оказывающих самостоятельное (не зависящее от природы жирных кислот липидов) действие на организм, также представлено в расчете на 100 г продукта.
Определение жиро кислотного состава с целью последующего количественного выражения в расчете на массу продукта возможно при наличии данных о фракционном составе липидов, так как жирные кислоты входят в состав ряда соединений (глицериды, свободные жирные кислоты, эфиры стеринов, фосфолипиды и др.). В каждой фракции соотношения жирных кислот и других компонентов (глицерин в глицеридах и фосфолипидах, стерины, аминоспирты) различны. Отражая состав жирных кислот в суммарных липидах, в целях последующего количественного выражения этих данных в пересчете на продукт, необходимо знать парциальные доли каждой фракции. Задача фракционирования липидов на основные классы соединений в настоящее время, как правило, решается с помощью адсорбционной хроматографии на силикагеле [2, 26].
Для количественного определения отдельных фракций адсорбционную хроматографию в тонких слоях селикагеля используют вместе с другими чувствительными методами анализа, например колориметрически или спектрофотометрически. После хроматографического разделения, обнаружения и идентификации компоненты элюируют растворителями из участков силикагеля, соответствующих отдельным фракциям [2], и определяют наиболее подходящим способом. Необходимо иметь в виду, что проведение хроматографического разделения таким способом всегда сопряжено с возможностью существенных потерь веществ на отдельных этапах. Поэтому необходимы тщательный контроль всех операций и, если возможно, сопоставление результатов определения индивидуальных соединений как по фракциям, так и по суммарным липидам.
Денситометрическое определение всех классов соединений на основе использования обычных методов проявления хроматограмм недостаточно точно ввиду отсутствия единой для всех соединений пропорциональности оптической плотности и концентрации. Однако оно менее трудоемко, чем при использовании экстракции. Исходя из этих положений, Д. И. Кузнецов и Л. И. Семенова [4] разработали методику денсито метрического определения индивидуальных классов липидов, в том числе стеринов, на основе хроматографии в тонком слое силикагеля. Принцип метода состоит в том, что перед анализом к силикагелю добавляют краситель метиловый красный. Липиды проявляются в виде пурпурных пятен на розовом, а затем желтом фоне. После обесцвечивания фона парами аммиака хроматограмму фотографируют, а фотокопии денситометрируют, сравнивая оптическую плотность изучаемых фракций с оптической плотностью стандарта.
Необходимо сделать некоторые общие замечания, которые следует учитывать при определении фракционного состава липидов. Для анализа используют суммарный липидный экстракт, предварительно освобожденный от нелипидных компонентов. Такая очистка предусмотрена в УСМВОЛ и приведена в описании метода [3] . При других методах экстракции, когда используют бинарные системы растворителей, экстракт, как правило, промывают слабыми водными растворами сильных электролитов (например, 0,87 %-ным раствором КСI) с последующим удалением верхней водной фазы, содержащей нелипидные примеси [21]. Может быть использована очистка на сефадексе G–25 [28]. Важно не допустить в процессе получения липидов продуктов их окисления, так как последние имеют иную хроматографическую подвижность, чем нативные липиды, и на хроматограммах будут присутствовать дополнительные пятна и "хвосты". Во избежание окисления липиды защищают от действия прямого солнечного света и хранят в холодильнике в плотно закрытых колбах (флаконах) с притертыми пробками, в которых находится экстракт. Растворители отгоняют в токе азота или под вакуумом, допуская лишь слабое (до температуры 40-50°С) нагревание. Выделенные для весового определения и подсушенные на воздухе липиды для фракционирования обычно не используют.
Для количественного определения отдельных классов липидов, главным образом триглицеридов, в некоторых случаях при наличии высокочувствительного денситометра используются методы тонкослойной хроматографии на готовых хроматографических пластинках "Силуфол" производства ЧССР (фольга с нанесенным тонким слоем силикагеля).
Хромато графические пластинки предварительно промывают от органических примесей, присутствующих в слое силикагеля, и пропитывают фосфорномолибденовой кислотой. Для этого пластинку помещают в ванночку с 1,0 %-ным раствором фосфорномолибденовой кислоты в смеси растворителей хлороформ-метанол (2:1) на 5 мин. В качестве растворителей можно использовать ацетон, этанол, четыреххло- ристый углерод и другие растворители, в которых растворяется фое- форномолибденовая кислота и органические примеси, присутствующие в слое силикагеля. Пластинку подсушивают на воздухе, а на линию старта (20 мм от края) наносят микрошприцем 1-2 мкл 5 %-ного хлороформенного раствора липидов (50-100 мкг) полосой 1 см. Пластинку помещают в хроматографическую камеру (желательно "сэндвич- камеру") и проявляют в смеси растворителей: гексан-диэтшювый эфир–уксусная кислота (90:2:1). Проявленную пластинку подсушивают на воздухе до исчезновения запаха растворителей и помещают в термостат с принудительной вентиляцией на 5 мин при температуре 60°С. В зависимости от конструкции термостата время и температура могут быть изменены. На светло-желтом фоне появляются темные пятна отдельных групп липидов. Интенсивность окраски пятен и их размеры определяют денситометрически.
ОПРЕДЕЛЕНИЕ СОСТАВА ЖИРНЫХ КИСЛОТ
МЕТОДОМ ГАЗОЖИДКОСТНОЙ ХРОМАТОГРАФИИ
Газожидкостная хроматография – единственный метод, который использовался для получения данных по жирнокис- лотному составу пищевых продуктов. Для анализа используют не сами жирные кислоты, а их производные – метиловые эфиры. Этим достигают высокую эффективность разделения при более низких температурах и более коротком времени анализа. При анализе должен быть использован метод, обеспечивающий количественный выход при превращении жирных кислот в метиловые эфиры. Предложен целый ряд таких методов [8, 23, 27] . Разработан также быстрый метод получения метиловых эфиров жирных кислот, использовавшийся в работах одного из авторов [11]. Метод прост и может быть рекомендован. Метанолиз глицеридов при использовании этого метода проходит очень быстро в закрытой системе в метанольном растворе КОН. Сильногидролизеванные липидные смеси подвергают кислотно-щелочному метанолизу [51.
Для получения метиловых эфиров можно использовать нагревание при 45-50°С в 5 %-ном растворе НС1 в абсолютном метаноле или 5– 10%-ном растворе толуолсульфокислоты в абсолютном метаноле. Для натуральных масел и жиров с кислотным числом меньше 2 для получения метиловых эфиров жирных кислот можно использовать метанолиз глицеридов в щелочной среде [8] .
Проведение газожидкостного хроматографического анализа в определенной степени зависит от типа хроматографа и колонок, а также от техники обработки хроматограмм (возможно использование автоматических расчетных устройств, прилагаемых к приборам). Ниже приведена общая аналитическая схема проведения анализа.
Газожидкостная хроматография метиловых эфиров жирных кислот может быть проведена как на набивных, так и на капиллярных колонках при условии получения хроматограмм, позволяющих осуществить количественный расчет содержания отдельных' компонентов смеси. Анализ проводят при температурном режиме для колонок в пределах 150-300°С.
В качестве газа-носителя используют азот, гелий или аргон, которые пропускают через колонку со скоростью 30-100 мл/мин. Метиловые эфиры детектируют и количественно определяют при помощи катарометра, ионизационного или пламенно-ионизационного детектора.
Хроматографическое разделение осуществляют, используя в качестве жидких полярных (при рабочей температуре) фаз полиэтилен-гликольадипат (LAC–1– Р–296), полипропиленгликольаднпат (реоплекс 400), бутандиолсукцинат и полиэшленгликольсукцинат, SP-1000, силары, OV-275 и неполярные SE-30, OV-101. При использовании капиллярных колонок рекомендуются полярные силиконовые фазы: OV-275 и силар-10С. Выбор фаз определяется конкретными задачами каждого исследования. Полярную жидкую фазу наносят на твердый носитель в количестве 5-15 %. Температура разделения на полярных фазах зависит от допустимой температуры работы фаз. Рекомендуется работать в режиме на 5-15°С ниже этой границы [1, 6] . Для идентификации используют сочетание данных хроматографического анализа, полученных при различных условиях (полярность жидкой фазы, состав твердого носителя, вид детектора, температура разделения), с данными, полученными нехроматографическими методами (окислительное расщепление, бромирование или каталитическое гидрирование двойных связей, спек тро фотометрия в ИК- или УФ-свете, масс-спектрометрия и т. д.).
Величины удерживаемых объектов Уд метиловых эфиров жирных кислот в значительной степени зависят от параметров разделения. Для идентификации отдельных компонентов смеси рекомендуется характеризовать их численным значением относительного удерживаемого объема VR, которое равно ошошению VR данного эфира к VR известного вещества – метилового эфира миристиновой, пальмитиновой или стеариновой кислоты [1].
Жирнокислотный состав большинства пищевых продуктов достаточно хорошо изучен, поэтому задача идентификации не сложная. Однако при изучении новых источников пищевых веществ и некоторых слабо обследованных объектов могут встретиться неидентифицированные соединения. В этом случае следует сообщать известное об их химической природе. Например, неидентифицирована кислота с 24 атомами углерода.
Для количеств ежой характеристики содержания жирных кислот определяют процентное отношение площади соответствующего пика хроматограммы к сумме площадей пиков.
Данные о составе метиловых эфиров жирных кислот используют затем для расчета содержания каждой жирной кислоты (в г на 100 г продукта). Расчет возможен, если имеются данные о фракционном составе изучаемого жира. Ниже рассматривается методика проведения такого расчета [5] .
РАСЧЕТ СОДЕРЖАНИЯ ИНДИВИДУАЛЬНЫХ
ЖИРНЫХ КИСЛОТ В ПИЩЕВЫХ ПРОДУКТАХ
Расчет содержания индивидуальной жирной кислоты (ее массы в граммах) в сумме липидов (жире), выделенной из пищевого продукта, а следовательно, и в целом пищевом продукте (по данным о содержании в нем жира) складывается из четырех этапов:
- расчет конверсионного фактора F;
- расчет содержания массы суммы жирных кислот в навеске анализируемого жира;
- расчет процентного содержания и массы индивидуальной жирной кислоты в анализируемом жире;
- расчет процентного содержания и массы индивидуальной жирной кислоты в пищевом продукте.
1. Конверсионный фактор F показывает, какая масса суммы жирных кислот приходится на единицу массы суммы липидов (жира). Поскольку в различных классах липидов на 1 моль липида приходится разное количество молей кислоты, необходимо ввести в расчетную формулу усредненный коэффициент ץ, показывающий соотношение молярных количеств гипотетического липида и содержащейся в нем гипотетической жирной кислоты:

Обычно с помощью газовой хроматографии получают величины относительного процентного содержания индивидуальных метиловых эфиров жирных кислот в сумме метиловых эфиров жирных кислот. И в ряде случаев, когда в анализируемом жире практически отсутствуют жирные кислоты, более короткоцепочечные, чем миристиновая кислота, десятичную долю жирной кислоты в смеси жирных кислот vi содержащихся в жире, можно заменить в уравнении (3) десятичной долей ее метилового эфира vi* в смеси метиловых эфиров этих же кислот, т. е. mi - mS vi*.
Однако при достаточно высоком содержании в жире короткоцепочечных жирных кислот, например в жире молока, молочных продуктов, сливочного масла, сыров, некоторых хлебобулочных изделий (сливочных сухарей) и т. п., такая замена недопустима. В этих случаях величину vi получают умножением десятичной доли метилового эфира индивидуальной кислоты vi на отношение молекулярной массы этой кислоты vi к молекулярной массе ее метилового эфира на отношение усредненной молекулярной массы метилового эфира гипотетической жирной кислоты μi*, представляемой данной суммой жирных кислот, к усредненной молекулярной массе гипотетической жирной кислоты μi:
![]()
Содержание индивидуальной жирной кислоты в жире (в г на 100 г или %)
![]()
Таким образом, для получения данных о содержании индивидуальной жирной кислоты в пищевом продукте необходимо предварительно определить содержание суммы липидов (жира) в пищевом продукте и особенно состав
лшшдов входящих в них жирных кислот для расчета значения конверсионного фактора F.
Американские специалисты при аналогичных расчетах используют приближенные значения F для целого ряда продуктов, основываясь главным образом на данных о содержании в жире триглицеридов и фосфолигшдов. Ряд таких значений приводится ниже [14, 16-20, 22, 24, 25, 29].
ПРИБЛИЖЕННЫЕ ЗНАЧЕНИИ КОНВЕРСИОННОГО ФАКТОРА
ДЛЯ ЛИПИДОВ РЯДА ПИЩЕВЫХ ПРОДУКТОВ


* Для жира рыб значение F приближенно рассчитывают по приводимой ниже диаграмме (19].
Количество отдельных жирных кислот в липидах можно определить и методом "внутреннего стандарта". В качестве внутреннего стандарта рекомендуется использовать кислоту, не содержащуюся в исследуемых липидах, например маргариновую. Однако при использовании метанолиза глицеридов в качестве внутреннего стандарта рекомендуется использовать не кислоты, а метиловые эфиры жирных кислот, например мётилмаргарат [8],
При анализе натурального жира или масла с содержанием триглицеридов более 97 % расчет количества жирных кислот рекомендуется проводить методом внутренней нормализации (т. е. когда все жирные кислоты суммируются и сумма кислот принимается за 100%, что численно равно общему содержанию липидов). Во всех остальных случаях расчет содержания отдельных жирных кислот липидов в пищевых продуктах проводят по формуле
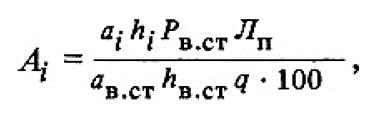

* Если анализ проводят в изотермическом режиме, то вместо ширины пика можно использовать время удерживания данного компонента. Если в комплекте прибора предусмотрен интегратор, то можно воспользоваться его показаниями.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ СТЕРННОВ
И ФОСФОЛИПИДОВ В ПИЩЕВЫХ ПРОДУКТАХ
Стерины и фосфолипиды обладают выраженной физиологической активностью и должны учитываться при расчете рационов питания, Большое практическое значение в настоящее время имеет вопрос о содержании в продуктах животного происхождения холестерина. При ряде нарушений липидного обмена (состояние гиперхолестеринемии) количество холестерина в суточном рационе нормируется, В продуктах растительного происхождения представляет интерес β-ситостерин, который является главным представителем растительных стеринов-фитостеринов, оказывающих гипохолестеринемическое действие. Как в продуктах животного, так и в продуктах растительного происхождения присутствует ряд других стеринов, но в меньших количествах. Физиологическая роль этих компонентов пока не выяснена. Достаточно знать содержание холестерина и |3-ситостерина. Для определения этих величин используют данные о содержании общих стериновых фракций, используя данные по холестерину для продуктов животного происхождения и по β-ситостерину – для растительных объектов.
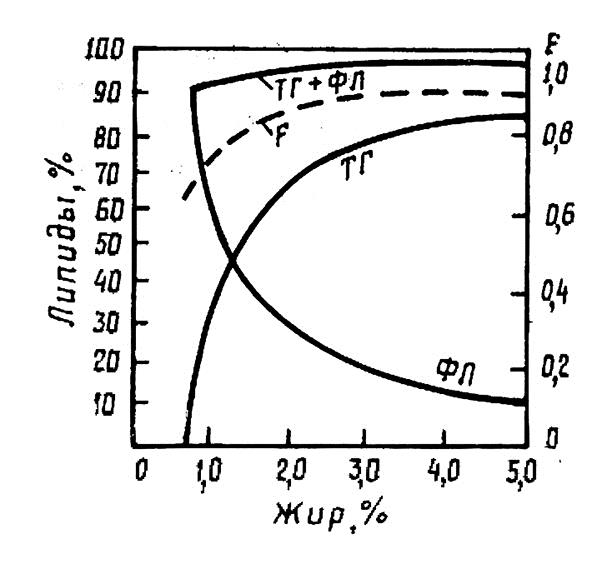
Диаграмма для расчета конверсионного фактора для жира рыб:
ТК - триглицериды; ФЛ - фосфоли пиды
Детальный анализ стериновых фракций может быть проведен с помощью газожидкостной хроматографии. Для этого необходимо предварительно выделить фракции неомыляемых веществ путем щелочного гидролиза [11] Общее содержание стеринов определяют колориметрически на основе цветных реакций. Для определения стеринов в пищевых продуктах, например, подходит цветная реакция с хлорным железом [30] .
При проведении анализа может быть использован суммарный липидный экстракт. Для одного анализа необходимо взять количество экстракта, содержащее не более 0,3 мг холестерина или β-ситостерина. Другие цветные реакции (например, Либермана–Бурхарда) менее пригодны, так как мешает окраска липидных экстрактов.
Определение содержания фосфолипидов осуществляется на основании анализа содержания липидного (липоидного) фосфора, т. е. фосфора, определяемого в экстракте липидов. Для этих целей можно использовать различные методы, так как во всех случаях липиды подвергаются минерализации. Существенным является лишь трудность минерализации образцов, в которых фосфолипиды составляют лишь небольшую долю в сравнении с триглицеридами. Часто для ускорения минерализации используют хлорную кислоту [2] , однако, увеличив время минерализации, можно применять и серную. Полученные величины содержания липидного фосфора умножают на усредненный лецитиновый коэффициент 25 и находят суммарное количество фосфолипидов.
* * *
Вариабельность данных по содержанию липидов и жирных кислот оценивалась по данным, представленным в МВК отраслевыми подкомиссиями при подготовке первого издания настоящего справочника [12] .
Вариабельность (среднеквадратичное относительное отклонение) общего содержания липидов оказалась довольно высокой [12]. Для большинства животных (кроме рыб) и растительных продуктов (кроме сои и овощей) эта величина находилась в пределах 10-15%. Для рыб, сои и овощей достигала 20-25 %. Это объясняется не только сортовыми или видовыми различиями, условиями выращивания, но в значительной степени методическими погрешностями.
Как отмечалось выше, для определения общих липидов используются различные методы, в том числе метод Фольча, метод Кузнецова и Гришиной, метод Сокслета и т. д. Межлабораторный коэффициент вариации различных методов составляет 7-10 %.
Еще большую вариабельность имеют данные по фракционному составу липидов, что в значительной степени объясняется разнообразием вариантов методов их определения и худшей межлабораторной сходимостью. В результате общая вариабельность данных по содержанию триглицеридов для большинства животных (кроме рыбы) и растительных продуктов находится в пределах 15-20%, для рыб 25-30 %. Для фосфолипидов (сумма), токоферолов и стеринов эти данные соответственно равны 10-15% (для большинства продуктов) и 20-25 % (для рыб). Для определения состава и количества жирных кислот, как отмечалось выше, используются исключительно методы газожидкостной хроматографии. Внутрилабораторная сходимость данных, полученных на современных хроматографах с набивными и капиллярными колонками, составляет 2 %. Межлабораторная воспроизводимость для большинства основных жирных кислот обычно в 2-3 раза выше. Вместе с тем следует помнить, что жирно кислотный состав продуктов зависит от сорта (вида), условий произрастания (содержания), хранения. Все это вместе взятое, а также естественное колебание в содержании общих липидов в продуктах приводит к тому, что общая вариабельность основных жирных кислот (тех, которые составляют более 10% относительно суммы жирных кислот) в большинстве продуктов составляет 15-20%, а в сое и рыбах – 30-35% [12].
Вариабельность минорных жирных кислот (1-10% суммы кислот) еще выше: для большинства продуктов 20-30%, а в сое и рыбе 35- 55% [12].


